



2026 年 4 月 9 日
The essential PK parameters and how to read them correctly
| wdt_ID | wdt_created_by | wdt_created_at | wdt_last_edited_by | wdt_last_edited_at | Parameter | Definition (plain language) | Mathematical intuition (very short) | Biological meaning (typical) | Development relevance | Common misconceptions |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | DaniellePillsbury | 09/04/2026 05:02 PM | DaniellePillsbury | 09/04/2026 05:02 PM | Cmax | Maximum observed concentration | Sample-based maximum; sensitive to early sampling | Proxy for peak systemic burden | BE endpoint; peak-driven safety concerns in some settings | “Higher Cmax always means better efficacy” (depends on exposure–response) |
| 2 | DaniellePillsbury | 09/04/2026 05:03 PM | DaniellePillsbury | 09/04/2026 05:03 PM | Tmax | Time to reach Cmax | Argmax time; unstable with sparse sampling | Absorption rate influenced timing | Often supportive (not always required statistically) | “Different Tmax always means non-equivalence” |
| 3 | DaniellePillsbury | 09/04/2026 05:04 PM | DaniellePillsbury | 09/04/2026 05:04 PM | AUC | Area under concentration–time curve | Integral (often trapezoids in NCA) | Overall systemic exposure | BE endpoint; basis for many dose/exposure discussions | “AUC tells you mechanism” (it’s integrative, not mechanistic) |
| 4 | DaniellePillsbury | 09/04/2026 05:05 PM | DaniellePillsbury | 07/05/2026 01:43 PM | Half-life (t½) | Time for concentration to halve in terminal phase | t½ = 0.693·V/CL (linear) | Joint signal of distribution + elimination | Washout, accumulation, dosing interval planning | “Long t½ means low clearance” (could also be high V) |
| 5 | DaniellePillsbury | 09/04/2026 05:06 PM | DaniellePillsbury | 09/04/2026 05:06 PM | Clearance (CL) | “Volume” cleared per time | CL = Rate_elim / C; also CL = F·Dose/AUC | Elimination capacity (integrating organs/processes) | Core for dose scaling and DDI/subpopulation impact | “CL is how much drug is removed per hour” (it’s a proportionality) |
| 6 | DaniellePillsbury | 09/04/2026 05:07 PM | DaniellePillsbury | 07/05/2026 01:44 PM | Volume of distribution (Vd) | Apparent volume relating amount to concentration | Vd ≈ Amount / C (conceptually) | Extent of distribution outside plasma | Loading dose logic; interpreting t½ shifts | “Vd is an anatomical volume” (it’s an apparent/theoretical one) |
Cmax: The peak plasma level a drug reaches
- Definition: Cmax (maximum observed concentration) is the highest measured drug concentration in the sampled concentration time profile, often in plasma or serum.
- Mathematical intuition: Cmax is a function of both the true peak and the sampling schedule. FDA explicitly notes that if the first sample is the highest point, Cmax may be biased due to insufficient early sampling.
- Biological meaning: Often a proxy for peak systemic burden; sometimes relevant for acute adverse events or rapid-onset pharmacology (context dependent).
- Development relevance: In EU BE, Cmax is a required parameter (with AUC) for single-dose studies.
- Common misconceptions: “Cmax is the efficacy metric.”E–R guidance stresses that clinical decisions need exposure–response understanding, not a single PK number.
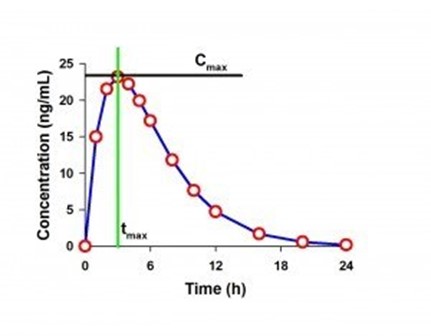
Fig.: Example PK Curve with Cmax and tmax labeled
Tmax: Capturing the speed of absorption
- Definition: Time to reach Cmax.
- Mathematical intuition: Tmax is discrete (depends on sampling times), making it noisy and sometimes non-symmetric statistically.
- Biological meaning: Influenced by absorption rate; in BE contexts, EMA notes Cmax and tmax are influenced by absorption rate.
- Development relevance: EMA states statistical evaluation of tmax is not required, but if rapid release is clinically important (onset or adverse events), there should be no “apparent” difference in median tmax and its variability.
- Common misconceptions: “Different Tmax always fails BE.”In many cases, it is supportive rather than primary.
AUC: Measuring total drug exposure over time
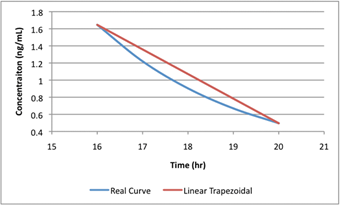
Fig.: Linear AUC assumes straight lines; real curves are not.
- Definition: Area under the concentration–time curve, i.e., total exposure over time.
- Mathematical intuition: AUC is an integral; in practice NCA uses trapezoidal approximations between observed points, plus a terminal extrapolation if estimating AUC∞.
- Biological meaning: In linear kinetics, AUC increases with dose and decreases with clearance.
- Development relevance: EMA specifies AUC(0–t) (or, when relevant, AUC(0–72h)) and Cmax for single-dose BE, with 90% CI acceptance 80.00–125.00%.
- Common misconceptions: “AUC captures absorption speed.”AUC is primarily an extent-of-exposure metric; rate can change Cmax/Tmax without changing AUC.
Half-life (t½): Understanding drug persistence over time
- Definition: Time for concentration to fall by half during the terminal phase (under linear/first-order assumptions).
- Mathematical intuition: t½ = 0.693/ke and, because CL = ke·Vd, also t½ = 0.693·Vd/CL.
- Biological meaning: Not a “pure elimination” metric – distribution (Vd) and elimination (CL) both matter.
- Development relevance: Guides washout length, sampling duration, and accumulation expectations for repeated dosing (regimen design).
- Common misconceptions: “Long half-life implies organ failure or low CL.”Half-life can increase if Vd increases even at constant CL.
Clearance (CL): The body’s capacity to eliminate drug
- Definition: “Rate of drug elimination divided by plasma concentration,” yielding an apparent plasma volume cleared per unit time.
- Mathematical intuition: In linear kinetics, CL links dose and exposure: AUC = F·Dose/CL (so CL = F·Dose/AUC).
- Biological meaning: Summarizes elimination rate across pathways (renal/hepatic/other).
- Development relevance: A primary driver of dose requirements and a key object/perpetrator concept in DDI workstreams (changes in CL often explain AUC changes).
- Common misconceptions: “CL tells you how much drug is removed per hour.”It is a proportionality; mg/h depends on concentration and CL.
Volume of distribution (Vd): How far a drug travels beyond the bloodstream
- Definition: An apparent/theoretical volume indicating the extent of distribution relative to plasma concentration.
- Mathematical intuition: Conceptually, Amount ≈ C·Vd (so Vd ≈ Amount/C).
- Biological meaning: A high volume of distribution typically indicates extensive distribution into tissues (or binding), not a “bigger body.”
- Development relevance: Influences loading-dose logic and helps interpret half-life changes (since t½ depends on Vd and CL).
- Common misconceptions: “Vd is anatomical volume.”It is explicitly described as apparent/theoretical.

Looking to go beyond the basics of PK parameters?
Authored by Prof. Johan Gabrielsson, the trusted reference book Pharmacokinetic and Pharmacodynamic Data Analysis provides comprehensive insights into pharmacokinetics, pharmacodynamics, and PK/PD concepts.
Register with your professional or academic email to access the full standard edition – completely free.
Learn more about Phoenix WinNonlin
Calculate and interpret PK parameters with confidence using Phoenix WinNonlin, industry trusted platforms that deliver validated noncompartmental analysis, modeling, and scalable cloud based workflows for modern drug development.


Sebastian Kuechenmeister
副总监Sebastian Küchenmeister joined Certara in 2022. He is a creative marketing professional with extensive expertise in multiple marketing disciplines, campaign management, media planning and a passion for content creation and go-to market strategies. Mr. Küchenmeister earned a Bachelor of Arts degree in Political Science from the Humboldt University in Berlin, Germany.
预约 Phoenix 演示
准备好见证 Phoenix 实战演示了吗?通过为您量身定制的指导演示,改变您的 PK/PD 分析。我们将向您展示 Phoenix 如何为您的药物开发之旅架起桥梁。




