


2026 年 3 月 20 日

Want to deepen your understanding of accumulation and other PK/PD concepts? Get free access to the 5th edition of Prof. Johan Gabrielsson’s renowned reference, Pharmacokinetic and Pharmacodynamic Data Analysis.
References and recommended reading
Downing NS, et al (2012). How Abbott’s Fenofibrate Franchise Avoided Generic Competition Arch Intern Med. 14; 172(9): 724–730.
Food and Drug Administration (1999). Center for Drug Evaluation and Research (CDER). Statistical Information from the June 1999 Draft Guidance and Statistical Information for In Vitro Bioequivalence Data, Posted on 1999 年 8 月 18 日. US Department of Health and Human Services, Rockville, MD.
Food and Drug Administration. Drug Approval Package: Tricor (fenofibrate) Tablets; Applications No.: 021203 and 021656.
Julious S et al (2010). An Introduction to Statistics in Early Phase Trials, John Wiley & Sons, NY.
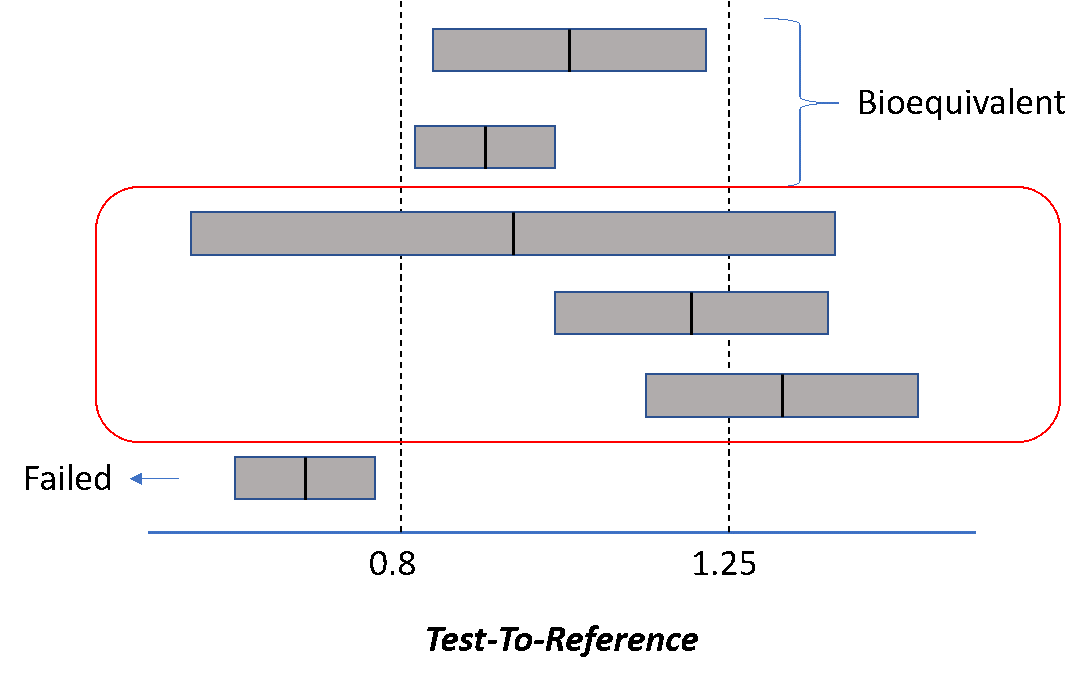
Food and Drug Administration (1992). Guidance for industry: statistical procedures for bioequivalence studies using a standard two treatment crossover design. US Department of Health and Human Services, Rockville, MD.
Food and Drug Administration (1995). SUPAC-IR: Immediate-Release Solid Oral Dosage Forms: Scale-Up and Post-Approval Changes: Chemistry, Manufacturing and Controls, In Vitro Dissolution Testing, and In Vivo Bioequivalence Documentation. US Department of Health and Human Services, Rockville, MD.
Food and Drug Administration (2001). Guidance for industry: statistical approaches to establishing bioequivalence. US Department of Health and Human Services, Rockville, MD.
Food and Drug Administration (2003). Guidance for industry: bioavailability and bioequivalence studies for orally administered drug products—general considerations. US Department of Health and Human Services, Rockville, MD.
Food and Drug Administration (2014). Guidance for industry: Bioavailability and Bioequivalence Studies Submitted in NDAs or INDs — General Considerations. US Department of Health and Human Services, Rockville, MD.
Krishna R, Yu L (2007). Biopharmaceutics applications in drug development, Springer, NY.
US Code of Federal Regulations. (2000). Bioavailability and bioequivalence requirements. US Government Printing Office, Washington, DC, Vol. 21, Part 320. Also see: https://www.govinfo.gov/content/pkg/CFR-2009-title21-vol5/pdf/CFR-2009-title21-vol5-sec320-1.pdf.
Williams RL et al (1976). Where are we now and where do we go next in terms of the scientific basis for regulation on bioavailability and bioequivalence? FDA Biopharmaceutics Coordinating Committee. European Journal of Drug Metabolism & Pharmacokinetics 25, 7 – 12.
常见问题解答
What software can I use to perform bioequivalence analyses?
Several programs, proprietary and non-proprietary, can be used to analyze data from BE studies. For example, Certara’s Phoenix WinNonlin® can be used to perform bioequivalence studies as well as reference-scaled average bioequivalence (RSABE). This is a statistical approach increasingly adopted to assess bioequivalence for highly variable drugs (HVDs).
What is virtual bioequivalence?
Virtual bioequivalence (VBE) refers to demonstrating bioequivalence using physiologically-based pharmacokinetic models instead of performing a clinical BE study. Many proprietary and non-proprietary software can run such VBEs, including Certara’s own Simcyp® Simulator.
If I get a similarity factor in dissolution studies of f₂ ≥ 50. what does that mean?
If your dissolution comparison gives f₂ ≥ 50, it means the test and reference products have similar dissolution profiles.
When Is VBE Most Valuable?
Virtual bioequivalence is most useful when:
- Clinical BE studies are expensive or risky.
- Dissolution data show small but uncertain differences.
- Regulatory justification needs strengthening.
- Multiple strengths require bridging.
- Ethical concerns limit in-vivo studies.

Rajesh Krishna, PhD
Senior Distinguished Scientist, Drug Development SolutionsRajesh is a scientific key opinion leader with 25+ years in drug development, specializing in model-informed strategies for biologics, vaccines, and small molecules. Currently a Senior Distinguished Scientist at Certara, he leads strategic consulting and the CDDS centers of excellence. Previously, he founded Merck’s quantitative clinical pharmacology department and held key roles at Aventis and Bristol-Myers Squibb. Rajesh holds a PhD in Pharmaceutical Sciences (University of British Columbia) and an MBA in Strategy and Innovation (Warwick). Consistently recognized among the top 2% of influential scientists, his work includes 100+ publications, 89 posters, and 4 books. He is an elected fellow of AAPS.
Contact us heading



