


How important is a clinical development plan?
Your biotechnology start-up’s candidate asset has an intriguing proof of biology. Your investors are equally excited about it, and everything looks rosy regarding your future investments. You have a draft target product profile (TPP) that is all developed and squared away.
Now, it is time to put your ideas into action! Admittedly, you run into some initial alarms. The patient population you want to recruit in your initial clinical trial is challenging. You’d be lucky to recruit 2-3 patients a month.
Now, you start to worry about your program. How will you manage idealistic vs realistic expectations? How will you balance executable vs impractical development scenarios?
听起来很熟悉吧?If yes, then this blog may be for you.
There are several ways to achieve focus in development. The one basic approach is to develop an early or clinical development plan (CDP). These can get sophisticated depending on how large your company is.
The CDP may be as simple as a table of pros and cons for each study scenario. Or it may run several hundred pages examining numerous scenarios and their pros and cons.
I’m an advocate for the former. Keep your CDP simple and use it as a living document. Adjust its associated assumptions as your candidate asset travels through its drug development journey.
The plan aims to develop a roadmap for the data required to support the TPP. At the minimum, it should detail the number and preliminary design of all planned clinical studies. In addition, it should articulate important scientific, medical, and regulatory submission considerations for asset registration.
Identifying the patient population, which impacts the target indication, is the first real value of the clinical development plan. Once you have identified the studies and the target population, the CDP can indicate your timeline’s practicality, costs, and required resources.
These aspects give decision-makers some sense of the regulatory landscape. They also provide an opportunity to interrogate your assumptions. If your company decides to add more assets to your portfolio, relative capitalization of the portfolio is achieved by developing these CDPs. These plans allow you to allocate your resources to the right project.
How to write a clinical development plan
The 3 key sections of any clinical development plan include
Clinical studies needed to achieve registration for the primary indication
Build on the TPP by distilling the clinical studies needed to support registration. A table of studies or an arrow diagram on a chart helps visualize how you will accomplish the following:
- Define clinical proof of biology,
- Identify the human dynamic range, and
- Initiate pivotal registration trials.
Here, you will identify preliminary synopses of studies, reflect on the questions answered, and the magnitude of changes expected. This information will then inform preliminary sample sizes and the data needed to either continue or discontinue the program.
Figure 1 below illustrates a hierarchy of endpoints.
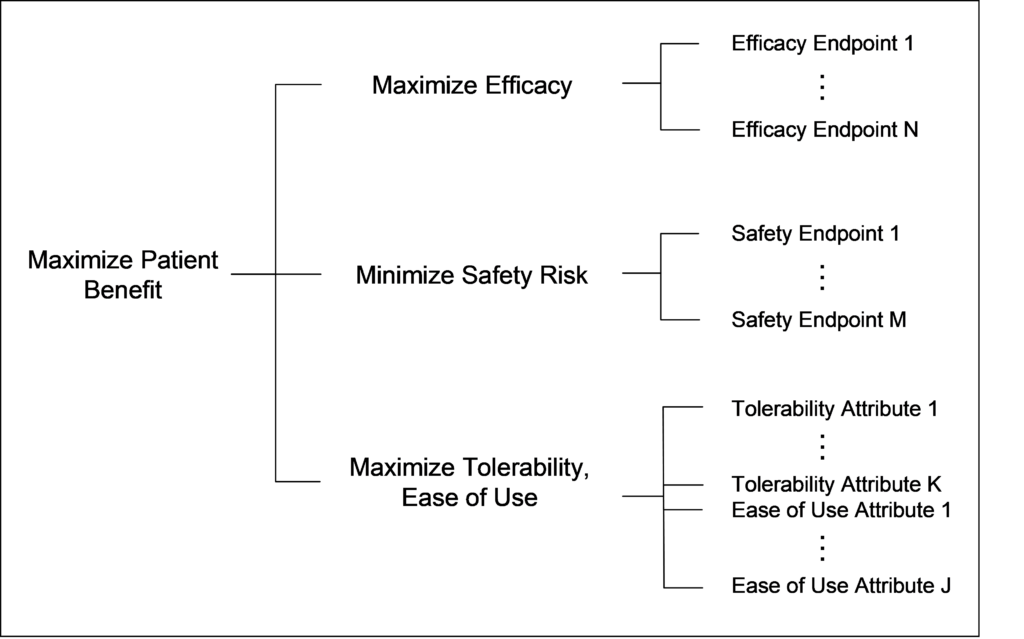
Valuable add-ons here include:
- A biomarker and surrogate endpoint strategy,
- Pediatric development strategy,
- Using model-informed drug development strategies to aid in clinical development decision-making or even conceptualize a blue-sky version of development (e.g., adaptive clinical trial designs or novel methods for dose selection and patient enrichment).
Likelihood of success or drivers of success for your program
The section builds out risks and failure points in your program so you can anticipate them and try to develop solutions upfront. Sponsors often stumble when they develop novel targets which lack current treatments. This is great from a blue-sky perspective but adds complexity to your execution plan.
Let’s say you are developing a glioblastoma therapy with a novel pharmacological target. This novelty means that it’s unclear what magnitude of clinical change is needed for a monotherapy vs in combination with standard of care.
Novel drug targets may not have a meaningful biomarker. The high risk identified allows you to understand potential alternatives. Perhaps this may lead you to engage key opinion leaders around your asset. Understanding the risk allows one to debate potential solutions to the issue.
One can apply modeling and simulation tools to better conceptualize key development questions and test potential scenarios. For a list of development questions by phase one can encounter, please see Figure 2.
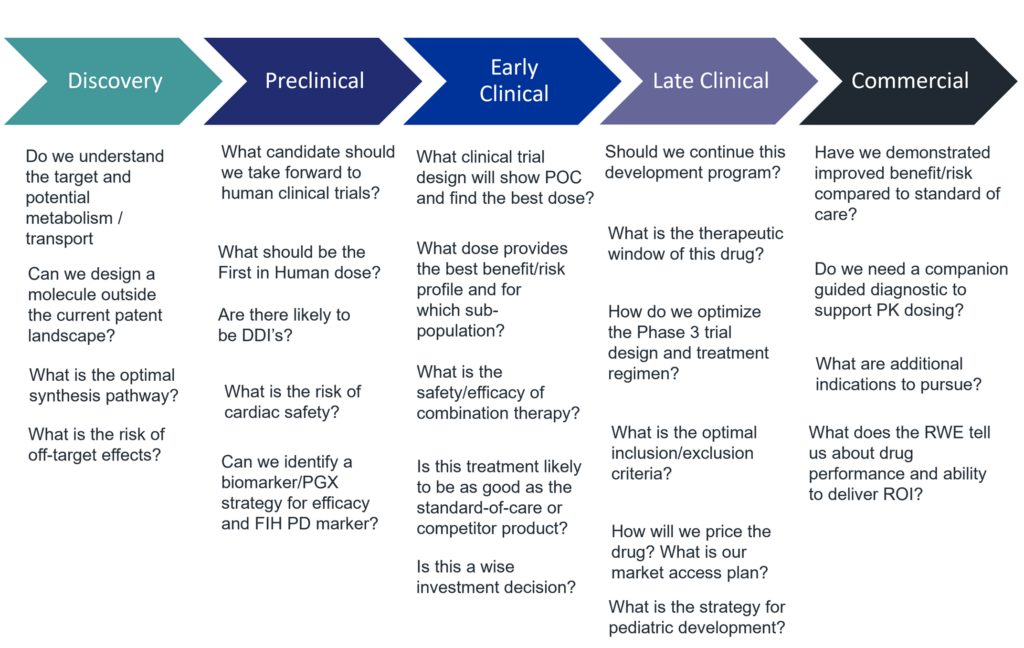
Regulatory requirements roadmap to maximize and integrate inputs
Understanding unique regulatory landscapes for your assets in the target areas of registration is vital. Often sponsors find quick and easy ways to start their first in human studies outside the US. However, they fall flat when they realize that their unique patient populations are not in the countries of interest. Whilst you aim to start your first in human study quickly, delays in your later studies can prolong your development timelines.
Your regulatory strategy is tied to your commercial strategy. If you are the only player in the field, your development path is simpler but higher risk. If you must compete against a standard of care situation, you are playing against competition. Figure 3 provides a conceptual framework for negotiating competition.
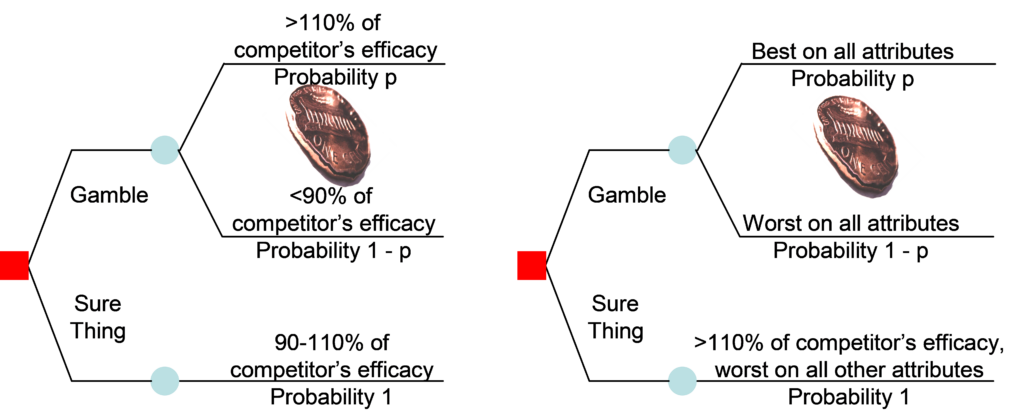
Read this blog for information about the importance of an Integrated Development Plan (IDP), including how an IDP relates to the Target Product Profile and the Clinical Development Plan (CDP).
I hope these reflections provide clues on how to develop ideation around your compound asset. Your asset has traveled this far. To realize its full potential, develop a dialog around the asset and whether it can pass regulatory muster.
The use of biomarkers is key to understanding drug mechanisms. It’s also a valuable part of a CDP. Read this white paper to learn why the inclusion and assessment of biomarkers within the drug development process can inform many critical decisions.
参考文献
- Poland et al. The Clinical Utility Index as a Practical Multiattribute Approach to Drug Development Decisions. Clin Pharmacol Ther. 2009 Jul;86(1):105-8.

