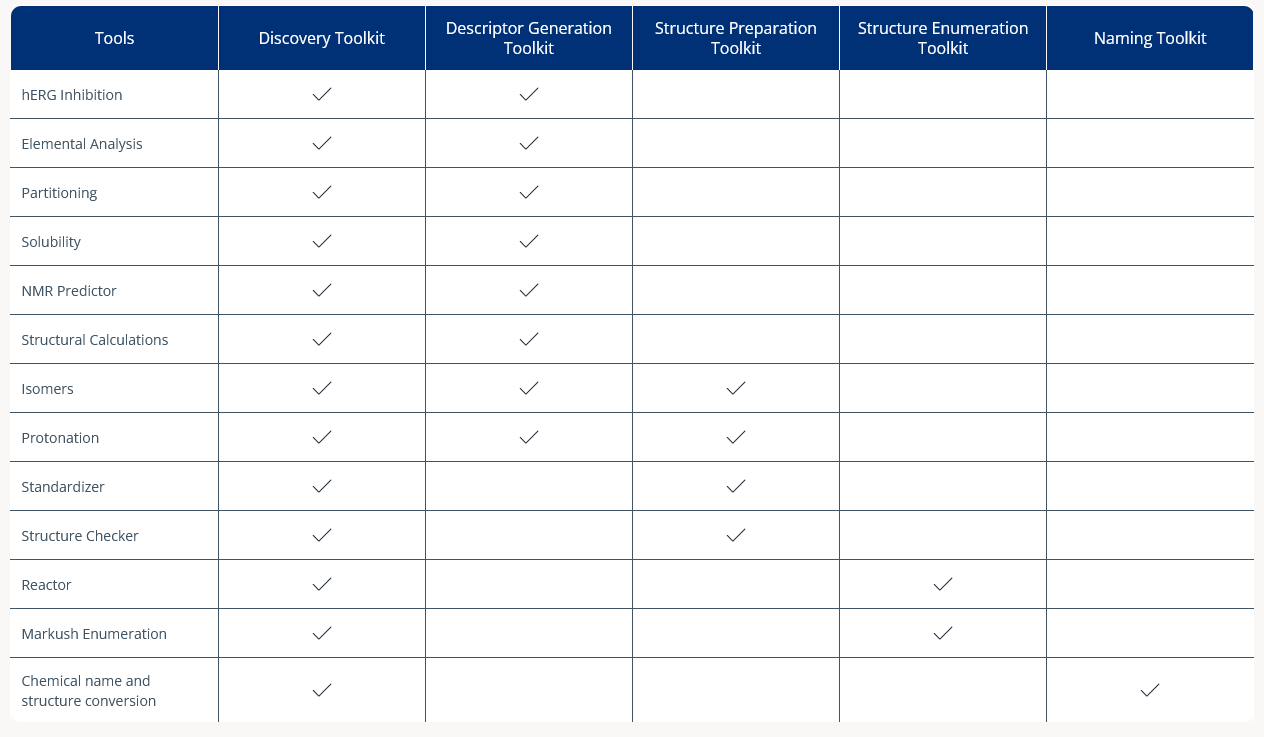
Assessing compounds in-silico is the most efficient way to prioritize new ideas, making sure only the best options are pursued and so minimizing effort spent on less promising compounds.
The Descriptor Generation Toolkit gives you the scientifically accurate property predictions needed to achieve this. It also allows you to generate the descriptors needed to thoroughly characterize the structures to train new models on your data.