


2025 年 9 月 5 日
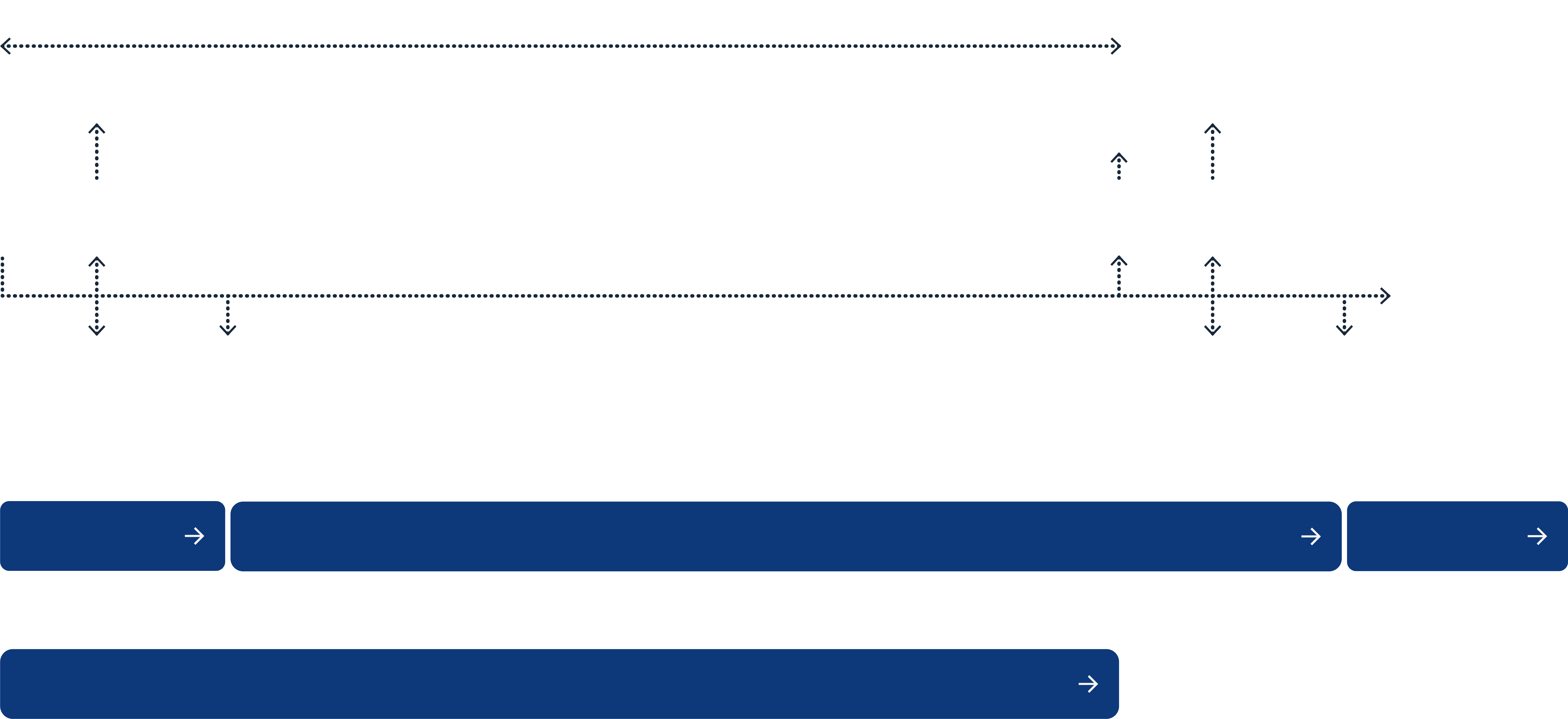

Figure 1: Example of the IB RSI update following the DSUR reporting period; adapted from CTR (EU) No. 536/2014 Q&A guidance document 2
Abbreviations: DSUR, Development safety update report; CTR, Clinical Trial Regulation; EU, European Union; IB, Investigator’s brochure; RSI, Reference Safety Information; SM, Substantial modification.
2. Regional requirements
The CTR (EU) No. 536/2014 Q&A guidance document also provides updated “Region-Specific Information” that is required in the DSUR submitted to the EU/European Economic Area (EEA) including:
- Cumulative summary tabulation of SARs
- List of subjects who died during the reporting period
- List of subjects who dropped out of clinical trials in association with an adverse event during the reporting period
- Safety signals review: additional information and appendix to present a high-level overview of the safety review process in the DSUR reporting period. Present the details and outcome of the safety signal review process during the DSUR reporting period in a table (like a periodic safety update report).
- EU clinical trial numbers of relevant trials should be listed alongside the protocol code.
Whilst the first 3 items listed above are consistent with the ICH E2F guidelines,3 items 2 and 3 have typically only been associated with regional appendices for the United States and are now a requirement for DSURs submitted to the EU/EEA. Item 4 aligns with the previously released guidance from the Medicines and Healthcare products Regulatory Agency/Health Canada5 in 2021. If signal evaluations for clinical trials are not possible/appropriate, then justification should be provided for not including the signal information. Item 5 is best addressed in Appendix 3 of the DSUR (Status of Ongoing and Completed Clinical Trials).
3. Removal of subject identifiers in data line listings
The CTR (EU) No. 536/2014 Q&A guidance document states that in order to comply with Article 43.3 of the CTR and protect patients’ rights, the line listing of SARs (in Appendix 5 of the DSUR) should identify SARs using the case identification (ID) and Study ID “without including subject ID,” and that the listing of subjects who withdrew from the study due to adverse events and the listing of subjects who died during the reporting period (refer to Section 2 above) should “not allow the identification of natural persons.”
Therefore, sponsors should not include clinical trial subject ID numbers in the line listings of SARs, the list of subjects who died during the reporting period, and the list of subjects who dropped out of studies during the reporting period due to adverse events.
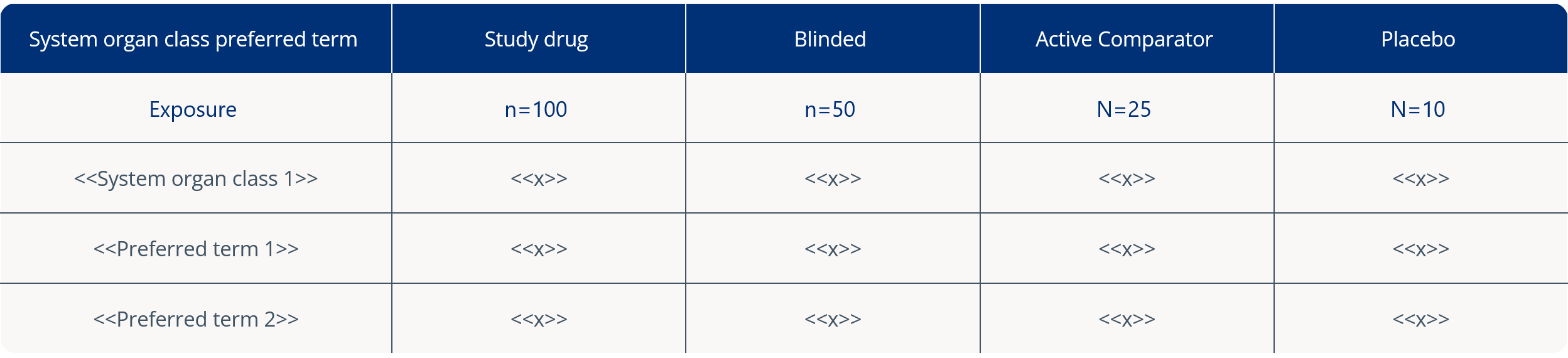
4. Cumulative summary tabulations of serious adverse events
The CTR (EU) No. 536/2014 Q&A guidance document suggests improving Section 7.3 of the DSUR ‘Cumulative Summary Tabulations of Serious Adverse Events’ by including the “absolute numbers of patients that have been treated.” This can be presented either within the body text of the DSUR or in the column headings of the Cumulative Summary Tabulation of Serious Adverse Events itself (in Appendix 6 of the DSUR), as illustrated below; adapted from the CTR (EU) No. 536/2014 Q&A guidance document.2

Adhering to regional guidelines to meet strict regulatory deadlines can be challenging. Certara regulatory writers are experts in authoring pharmacovigilance documents, and harnessing their expertise is a great asset to any biopharmaceutical company. For more information on pharmacovigilance documents, read this white paper.
参考文献
- Clinical Trials Regulation (Regulation (EU) No. 536/2014). Available from: https://www.ema.europa.eu/en/human-regulatory-overview/research-development/clinical-trials-human-medicines/clinical-trials-regulation.
- The rules governing medicinal products in the European Union volume 10 – Guidance documents applying to clinical trials. Clinical Trials Regulation (EU) NO 536/2014. Questions and Answers. Version 7.1. Accessed on 29 April 2025. Available from: https://health.ec.europa.eu/latest-updates/questions-and-answers-document-regulation-eu-5362014-version-5-january-2022-2022-02-01_en.
- International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. ICH harmonised tripartite guideline: Development safety update report E2F. Accessed on 29 April 2025. Available from: https://www.ema.europa.eu/en/documents/scientific-guideline/ich-guideline-e2f-development-safety-update-report-step-5_en.pdf.
- Clinical Trial Facilitation Group (CTFG) Q&A document – Reference safety information Accessed on 29 April 2025. Available from: https://www.hma.eu/fileadmin/dateien/Human_Medicines/01-About_HMA/Working_Groups/CTFG/2017_11_CTFG_Question_and_Answer_on_Reference_Safety_Information_2017.pdf.
- Guideline on how to increase transparency when presenting safety information in the Development Safety Update Report (DSUR): region-specific requirements for Canada and the United Kingdom. Last accessed 29 April 2025. Available from: https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/993808/DSUR-Guideline-08June2021.pdf.

Principal Regulatory Writer
Dr. Nicholas Churton is a principal regulatory writer with over 10 years of experience working in a clinical research organization. He has authored multiple regulatory documents across various therapeutic areas and multiple phases, specializing in aggregate safety reporting documents such as development safety update reports (DSURs), periodic benefit-risk evaluation reports (PBRERs), risk management plans (RMPs), periodic adverse drug experience reports (PADERs), and responses to health authority queries. He has experience in managing large cross-functional teams on pivotal projects in addition to developing training resources and providing oversight of junior writers.
常见问题解答
What is the EU Clinical Trials Regulation (CTR) 536/2014, and how does it impact my annual safety reports?
The EU CTR No. 536/2014, which replaced the Clinical Trials Directive in January 2022, harmonizes clinical trial processes across the European Union (EU). It introduces additional requirements for annual safety reports (ASRs), also known as development safety update reports (DSURs), including updated Reference Safety Information (RSI) alignment, enhanced regional requirements, and modified data presentation standards.
How should I handle RSI updates under the new regulation?
Under EU CTR No. 536/2014, the RSI section of your Investigator’s brochure should be updated annually to align with your DSUR data lock point and submitted within one month of the DSUR. This updated RSI becomes effective for the subsequent reporting period, ensuring consistency in expectedness assessments for serious adverse reactions.
What new regional requirements must be included in EU/EEA DSUR submissions?
The CTR Q&A guidance document requires five key regional elements: a cumulative summary tabulation of serious adverse reactions (SARs), lists of subjects who died during the reporting period, lists of subjects who withdrew due to adverse events, a comprehensive safety signal review with tabulated outcomes, and inclusion of EU clinical trial numbers alongside protocol codes.
How has subject privacy protection changed under the new regulation?
To comply with Article 43.3 of the CTR and protect patient rights, you must remove all subject identifiers from data line listings. This includes using only case identification numbers and Study IDs in SAR listings, while ensuring that death and withdrawal listings cannot identify natural persons.
联系我们



